The general SAM workflow contains several checkpoints (depicted with traffic-light symbols). At each checkpoint, every compound is evaluated and is either progressed along, or is eliminated from, the list of compounds. For example, after the binding affinity prediction step only those compounds that are predicted to be high-affinity binders (and thus should be good inhibitors of the target protein) are subject to absorption, distribution, metabolism, and excretion (ADME) and Toxicity predictions. Subsequently, only those compounds with favorable ADME predictions and absence of toxicities are subject to the binding affinity assay, and so on.
A detailed storyboard was developed to guide the initial drug design activities of SAM, which focused on obtaining consensus predictions of activities from different methods which could be used for compound prioritization for testing. This storyboard both demonstrates the basic modeling principles used in such design activities and the use cases that need to be supported by collaborative infrastructure and services.
One of the first activities of SAM was to identify a protein target whose function is essential to the survival of Plasmodium. It was agreed to focus on the kinase family of proteins. Cristian Doerig suggested several kinases whose proper functions are known from reverse genetics data to be essential for the malarial parasite. These suggested kinases were evaluated for their feasibility for SAM.
For this evaluation, several factors were important:
1) The availability/existence of an experimental activity assay.
2) The availability of structural information on the protein target (three-dimensional coordinates of all atoms, either experimentally determined or computationally modeled).
3) Knowledge of specific ligands/inhibitors.
4) Potential for selectivity (i.e., sufficiently large difference of interaction from that of Q2 any human kinase).
5) Novelty (the kinase should not already have been exploited in drug discovery research).

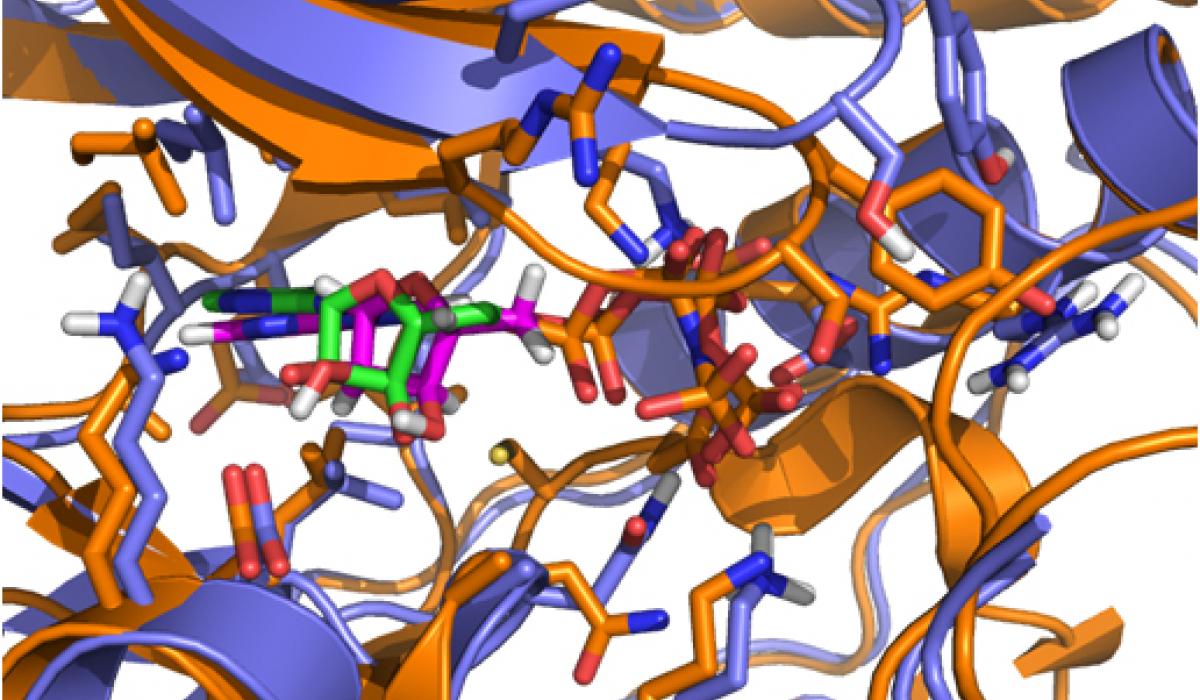
While CD investigated the availability of an experimental assay, RA analyzed the proposed kinases according to the remaining factors. Eventually, the selection came down to two kinases: one kinase offered the possibility of obtaining high-quality model coordinates, but appeared to be problematic in terms of allowing for selectivity; the second kinase proved to be a very novel target with good potential for selectivity, but was problematic in terms of obtaining accurate model coordinates (no crystal structure was available for it when the VO was initiated). In a virtual meeting, it was decided to select the latter kinase, a member of the mitogenactivated protein kinase (MAPK) family called Pfmap-2. Later, this choice turned out to be a good one, when the structures of new malarial kinases were acquired. It also emerged that these new structures allowed accurate model coordinates for Pfmap-2 to be created based on homology modeling. This modeling activity was done by RA, and the model was subsequently refined by AC and HGT. The Pfmap-2 model produced is shown in the Figure, and was subsequently used for pharmacophore and docking-based library design. During the operation of the VO, the Structural Genomics Consortium (SGC) published the crystal structure of Pfmap-2, and it was rewarding to see that the model was a close prediction of the crystal structure.
UC DDC had a physical library of approximately 300 000 chemicals that all have the general characteristics (size, chemical properties) of drug-like molecules, and they made knowledge on this library available for SAM. Many of these molecules interconvert rapidly between several physico-chemical states.